PLX038
PLX038 (PEG~SN-38) – A long-acting DNA damaging agent designed for use in cancers with a defective DNA damage response (DDR) and inhibitors of the DDR.
PLX038 (PEG~SN-38) is a long-acting PEGylated prodrug of SN-38, the active metabolite of the approved anti-cancer agents irinotecan (Camptosar®, CPT-11) and Trodelvy® (sacituzumab govitecan). SN-38 is a potent inhibitor of topoisomerase 1 (topo1) that causes replication stress, inhibition of DNA synthesis and DNA damage leading to cell death in susceptible tumors. As a long-acting DNA damaging agent, PLX038 causes synthetic lethality in tumors with DNA damage repair defects and enhances the effects of drugs that inhibit the DNA damage response (DDR), such as PARP inhibitors.
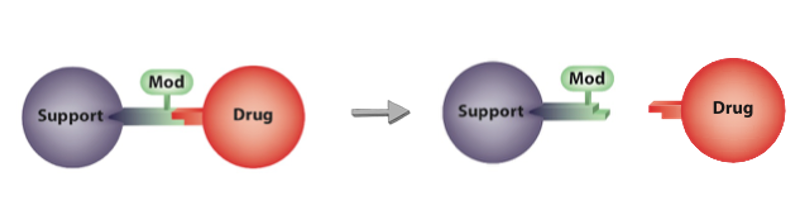
The long-acting PLX038 was prepared by coupling SN-38 (Drug) to the large carrier PEG (Support) with proprietary linkers that permit the controlled, slow release of SN-38.
PLX038 has been evaluated in a complete Phase I clinical trial at M.D. Anderson Center and is currently being studied, alone or in combination with PARP and ATR inhibitors, in several ongoing Phase I/II trials, including in patients with platinum resistant ovarian cancer, malignant CNS tumors, triple negative breast cancer and other advanced solid tumors. One objective of these trials is to identify biomarkers that will be useful in selection of patients mostly likely to respond to PLX038 therapy.
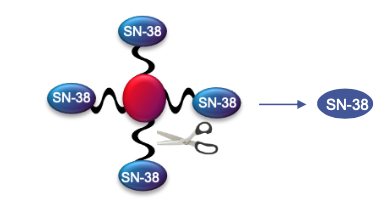
The long-acting PLX038 contains four SN-38s releasably connected to a 4-arm 40 kDa PEG through ProLynx linkers, and slowly releases SN-38 in the physiological environment.
Topoisomerase 1 Inhibition. Inhibition of Topo1 leads to single-strand DNA breaks and leads to replication stress, inhibition of DNA synthesis, and ultimately cell death in susceptible tumors. It has been shown that continuous, protracted administration of topo1 inhibitors is advantageous over intensive bolus-delivery. It appears that a minimum threshold of exposure to a topo1 inhibitor over a long period is advantageous for increased efficacy and reduced toxicity; higher dose-intensity enhances toxicity but not tumor regression.
SN-38. SN-38 is one of the most potent inhibitors of topo1. SN-38 is too insoluble and has too short of a half-life to administer directly, and requires the use of soluble pro-drugs. Irinotecan, the paradigm SN-38 prodrug, is FDA approved and primarily used for treatment of colon cancer in combination with other agents. While highly effective, Irinotecan has many undesirable features:
- It is extensively metabolized so its pharmacokinetics and pharmacodynamics show high inter-patient variability.
- It is administered as a bolus infusion and shows a high Cmax – associated with toxicity – and short half-life.
- It requires activation to SN-38 in the liver, and as a consequence often shows severe GI toxicity due to extensive glucuronidation and excretion into the bile.
- The high toxicity and short half-life limit dosing regimens such that topo1 is not effectively inhibited for most of the dosing cycle.
Irinotecan is thus a sub-optimal prodrug of SN-38, with limited effectiveness as an antitumor therapeutic.
DNA Damage Response. Cells undergo thousands of DNA damage events each day. However, normal cells mount an effective “DNA damage response” (DDR) to recognize and correct the DNA damage with high fidelity, even in the face of DNA damaging agents such as topo1 inhibitors. However, many cancers have DDR defects and are less able to repair DNA damage, such as the situation with BRCA-deficiency. Also, even if tumor cell viability is conserved with one DDR defect, impairment of two or more – for example, in a tumor with a genetic BRCA deficiency treated with a PARP inhibitor – results in cell death by synthetic lethality. Cancers having a defective DDR display heightened sensitivity towards DNA-damaging agents – in particular PLX038 – due to a decreased ability to repair DNA damage resulting in lethality. Because of its superior pharmacokinetics, PLX038 has the potential to be used in combination with a variety of DDR inhibitors and in tumors which have pre-existing DDR defects. Tumors with a weakened DDR comprise a substantial portion of cancers and can be detected with use of appropriate biomarkers.
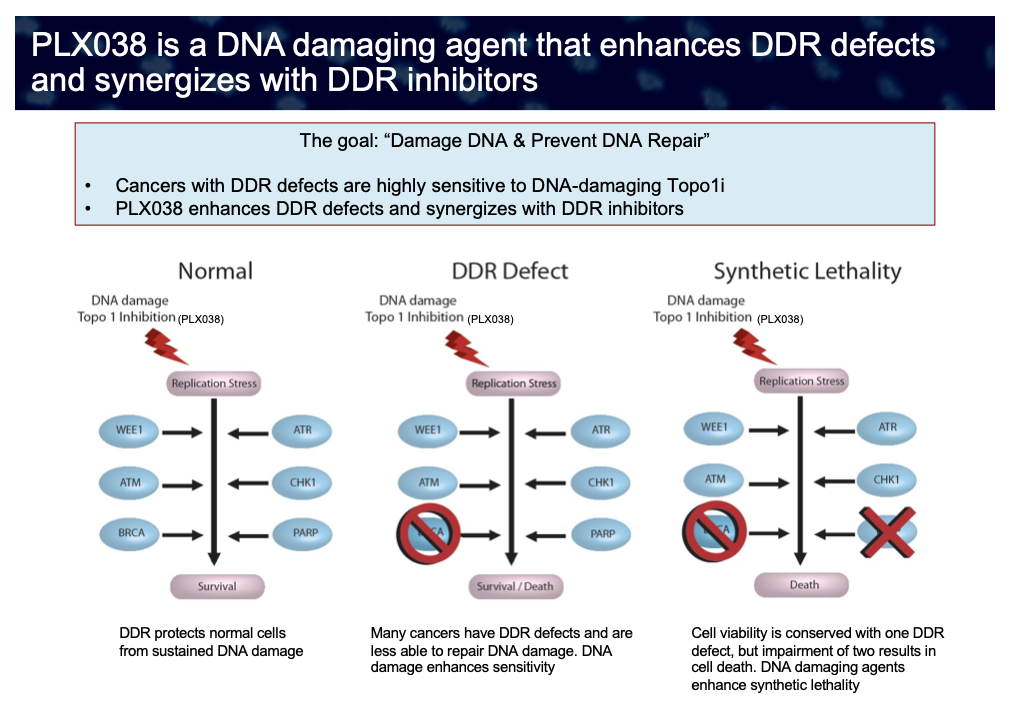
Defects in the DNA damage response have been recognized in many different cancer types. As a result, inhibitors of components of the DDR response which could cause synthetic lethality in such cells – such as PARP and ATR inhibitors – have assumed high importance in treatment of cancer. As such, numerous inhibitors of the DDR are in development, and it may be speculated that a single DNA damaging agent – such as PLX038 – might enhance the anti-tumor effects of many or all of them.
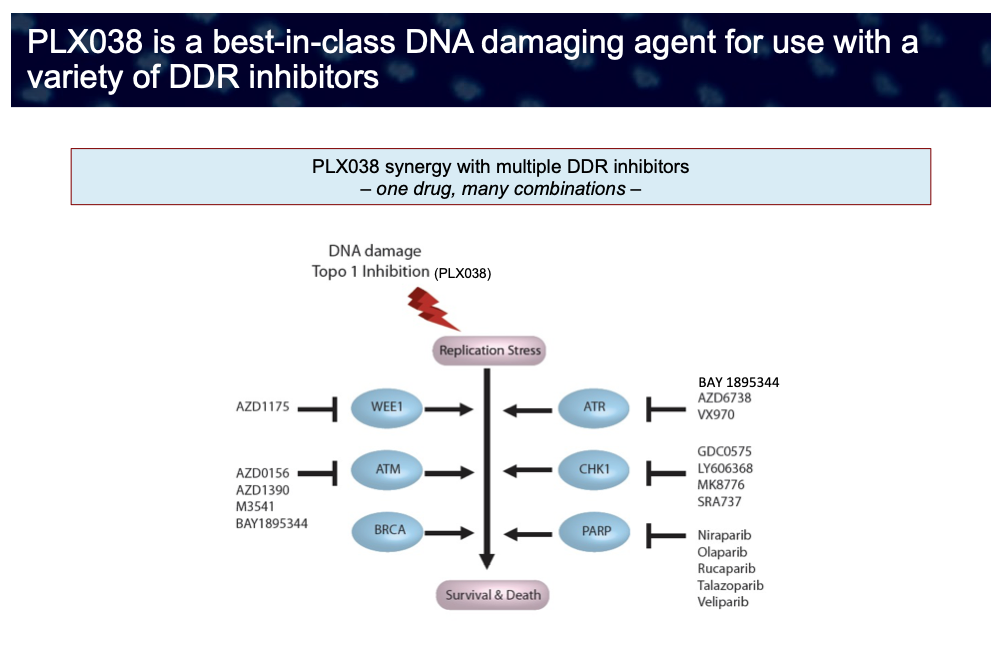
Thus, DNA damaging agents can be effective in (i) tumors with germline or somatic DDR defects – such as cells deficient in BRCA1/2, ATM or other DNA homologous recombination repair defects (i.e., cells with “BRCAness”), – and (ii) in cells where DDR deficiency is induced through chemical means, for example, in combination therapy with inhibitors of various DDR components, e.g., PARP, CHK1, ATM or ATR inhibitors.
PLX038 shows remarkable antitumor effects in preclinical models of human tumors with DDR defects and those treated with DDR inhibitors. For example, in the BRCA1-deficient human xenograft MX-1, a single, non-toxic dose of PEG~SN-38 completely inhibited tumor growth and resolved massive tumors.
PLX038 Differentiation and use with DDR inhibitors. PLX038 was designed to address the liabilities of Irinotecan and thus provide a superior anti-tumor agent. PLX038 exhibits a unique combination of the most desirable features of a topo1 inhibitor: (i) released SN- 38 has a very long half-life (5 days in human) and consequently a very low Cmax; (ii) SN- 38 is non-enzymatically released in the plasma rather than enzymatically in the liver, and so shows much lower glucuronidation and GI toxicity than Irinotecan; (iii) PLX038 accumulates in solid tumors, and (iv) shows high therapeutic efficacy.
We have now developed PLX038 such that it is the best-in-class DNA damaging agent for use with DDR inhibitors including, but not limited to, the approved PARP inhibitors, and inhibitors of specific components of the DDR (e.g., ATR, CHK1, Wee1 kinase) now in development. Because of its long duration of DNA damage (~2 out of a 3 week cycle) compared to CPT-11 (~12 hr of a 3 week cycle), it provides a long time-window within which the DDR inhibitor may be optimally scheduled to achieve synthetic lethality.